
Un nouvel outil d’IA classe les effets de 71 millions de mutations « faux-sens »
Découvrir les causes profondes des maladies constitue l’un des plus grands défis de la génétique humaine. Avec des millions de mutations possibles et des données expérimentales limitées, le mystère reste largement de savoir lesquelles pourraient donner naissance à la maladie. Ces connaissances sont cruciales pour accélérer le diagnostic et développer des traitements salvateurs.
Aujourd’hui, nous publions un catalogue de mutations « faux-sens » dans lequel les chercheurs peuvent en apprendre davantage sur leurs effets possibles. Les variantes faux-sens sont des mutations génétiques qui peuvent affecter la fonction des protéines humaines. Dans certains cas, elles peuvent entraîner des maladies telles que la mucoviscidose, la drépanocytose ou le cancer.
Le catalogue AlphaMissense a été développé à l’aide d’AlphaMissense, notre nouveau modèle d’IA qui classe les variantes faux-sens. Dans un article publié dans Science, nous montrons qu’il a classé 89 % des 71 millions de variantes faux-sens possibles comme étant probablement pathogènes ou probablement bénignes. En revanche, seuls 0,1 % ont été confirmés par des experts humains.
Les outils d’IA capables de prédire avec précision l’effet des variantes ont le pouvoir d’accélérer la recherche dans des domaines allant de la biologie moléculaire à la génétique clinique et statistique. Les expériences visant à découvrir des mutations pathogènes sont coûteuses et laborieuses : chaque protéine est unique et chaque expérience doit être conçue séparément, ce qui peut prendre des mois. En utilisant les prédictions de l’IA, les chercheurs peuvent obtenir un aperçu des résultats pour des milliers de protéines à la fois, ce qui peut aider à hiérarchiser les ressources et à accélérer des études plus complexes.
Nous avons mis toutes nos prédictions gratuitement à la disposition de la communauté des chercheurs et ouvert le code modèle pour AlphaMissense.
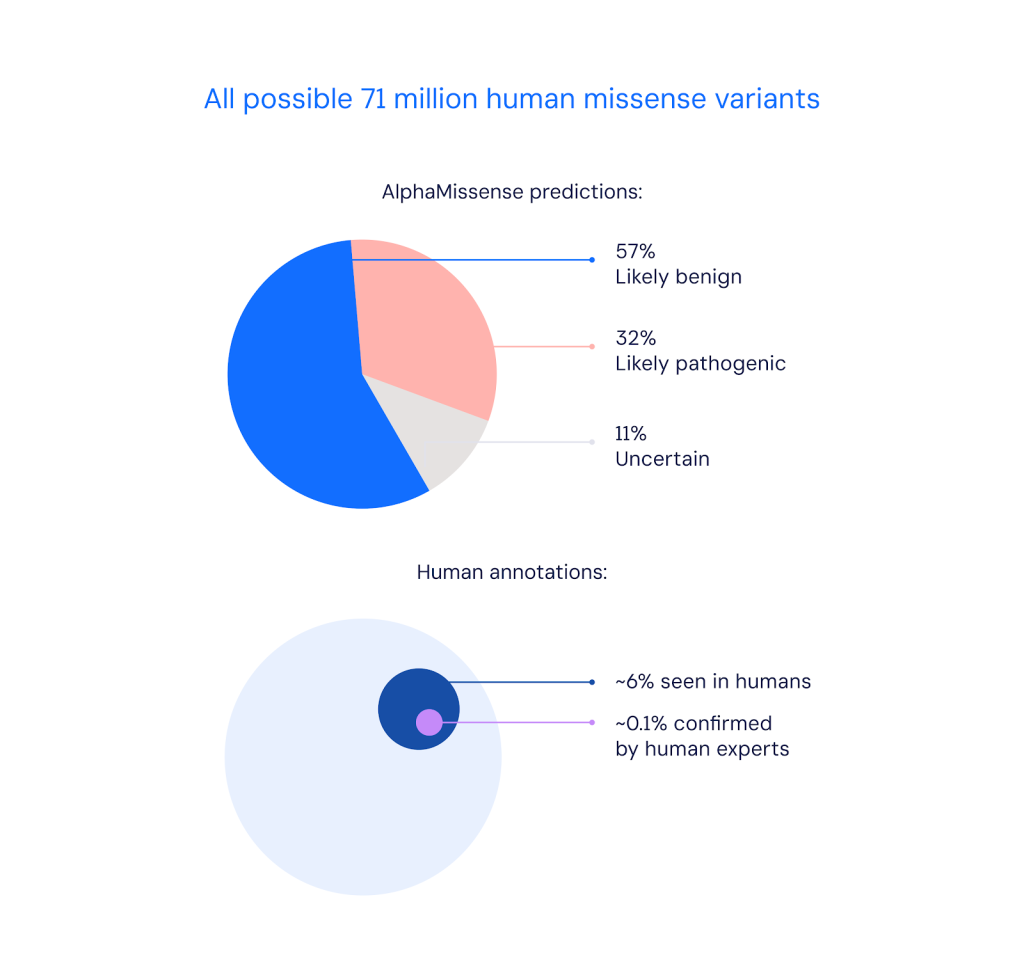
AlphaMissense predicted the pathogenicity of all possible 71 million missense variants. It classified 89% – predicting 57% were likely benign and 32% were likely pathogenic.
Qu’est-ce qu’une variante faux-sens ?
Une variante faux-sens est une substitution d’une seule lettre dans l’ADN qui aboutit à un acide aminé différent dans une protéine. Si vous considérez l’ADN comme un langage, changer une lettre peut changer un mot et modifier complètement le sens d’une phrase. Dans ce cas, une substitution modifie l’acide aminé traduit, ce qui peut affecter la fonction d’une protéine.
Une personne moyenne est porteuse de plus de 9 000 variantes de faux-sens. La plupart sont bénignes et ont peu ou pas d’effet, mais d’autres sont pathogènes et peuvent gravement perturber la fonction des protéines. Les variantes faux-sens peuvent être utilisées dans le diagnostic de maladies génétiques rares, où quelques variantes faux-sens, voire une seule, peuvent directement provoquer une maladie. Ils sont également importants pour étudier des maladies complexes, comme le diabète de type 2, qui peuvent être provoquées par une combinaison de nombreux types différents de modifications génétiques.
La classification des variantes faux-sens est une étape importante pour comprendre lesquels de ces changements protéiques pourraient donner lieu à une maladie. Sur plus de 4 millions de variantes faux-sens déjà observées chez l’homme, seulement 2 % ont été annotées comme pathogènes ou bénignes par les experts, soit environ 0,1 % des 71 millions de variantes faux-sens possibles. Les autres sont considérés comme des « variantes d’importance inconnue » en raison du manque de données expérimentales ou cliniques sur leur impact. Avec AlphaMissense, nous disposons désormais de l’image la plus claire à ce jour en classant 89 % des variantes en utilisant un seuil qui a donné une précision de 90 % sur une base de données de variantes de maladies connues.
Pathogène ou bénin : comment AlphaMissense classe les variantes
AlphaMissense est basé sur notre modèle révolutionnaire AlphaFold, qui prédit les structures de presque toutes les protéines connues de la science à partir de leurs séquences d’acides aminés. Notre modèle adapté peut prédire la pathogénicité des variantes faux-sens altérant les acides aminés individuels des protéines.
Pour former AlphaMissense, nous avons affiné AlphaFold sur des étiquettes distinguant les variantes observées dans les populations humaines et de primates étroitement apparentées. Les variantes couramment observées sont traitées comme bénignes et les variantes jamais observées sont traitées comme pathogènes. AlphaMissense ne prédit pas le changement dans la structure des protéines lors d’une mutation ou d’autres effets sur la stabilité des protéines. Au lieu de cela, il exploite des bases de données de séquences protéiques associées et le contexte structurel des variantes pour produire un score compris entre 0 et 1, évaluant approximativement la probabilité qu’une variante soit pathogène. Le score continu permet aux utilisateurs de choisir un seuil de classification des variantes comme pathogènes ou bénignes qui correspond à leurs exigences de précision.
AlphaMissense réalise des prédictions de pointe sur un large éventail de références génétiques et expérimentales, le tout sans formation explicite sur ces données. Notre outil a surpassé les autres méthodes informatiques lorsqu’il est utilisé pour classer les variantes de ClinVar, une archive publique de données sur la relation entre les variantes humaines et la maladie. Notre modèle était également la méthode la plus précise pour prédire les résultats du laboratoire, ce qui montre qu’il est cohérent avec différentes manières de mesurer la pathogénicité.
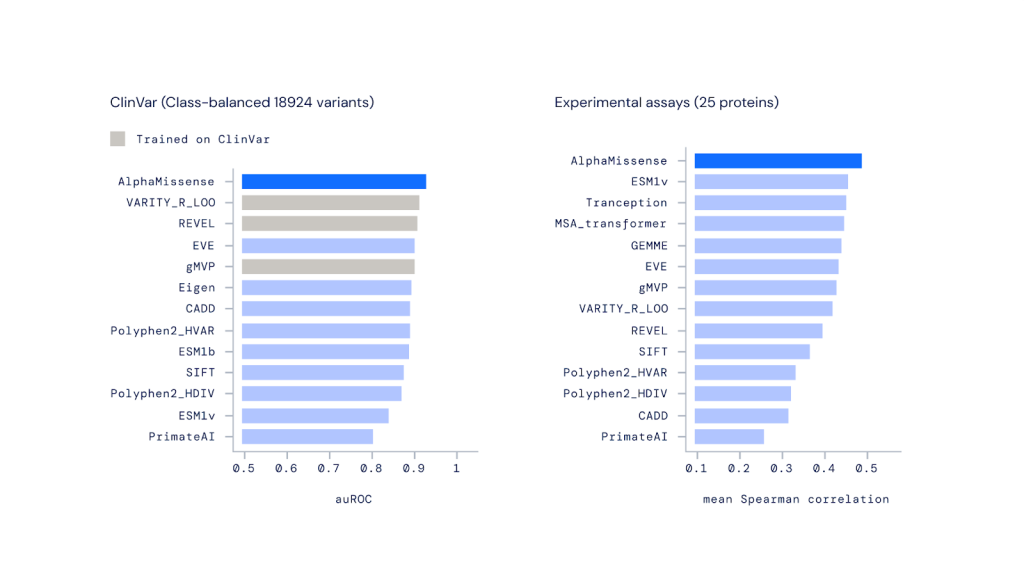
AlphaMissense outperforms other computational methods on predicting missense variant effects.
Left: Comparing AlphaMissense and other methods’ performance on classifying variants from the Clinvar public archive. Methods shown in grey were trained directly on ClinVar and their performance on this benchmark are likely overestimated since some of their training variants are contained in this test set.
Right: Graph comparing AlphaMissense and other methods’ performance on predicting measurements from biological experiments.
Construire une ressource communautaire
AlphaMissense s’appuie sur AlphaFold pour approfondir la compréhension mondiale des protéines. Il y a un an, nous avons publié 200 millions de structures protéiques prédites grâce à AlphaFold, ce qui aide des millions de scientifiques du monde entier à accélérer la recherche et à ouvrir la voie à de nouvelles découvertes. Nous sommes impatients de voir comment AlphaMissense peut aider à résoudre des questions ouvertes au cœur de la génomique et dans les sciences biologiques.
Nous avons mis les prédictions d’AlphaMissense à la disposition gratuite de la communauté scientifique. En collaboration avec l’EMBL-EBI, nous les rendons également plus utilisables pour les chercheurs grâce à l’Ensembl Variant Effect Predictor.
En plus de notre table de recherche des mutations faux-sens, nous avons partagé les prédictions élargies de toutes les substitutions possibles de 216 millions de séquences d’acides aminés uniques sur plus de 19 000 protéines humaines. Nous avons également inclus la prédiction moyenne pour chaque gène, ce qui est similaire à la mesure de la contrainte évolutive d’un gène : cela indique à quel point le gène est essentiel à la survie de l’organisme.
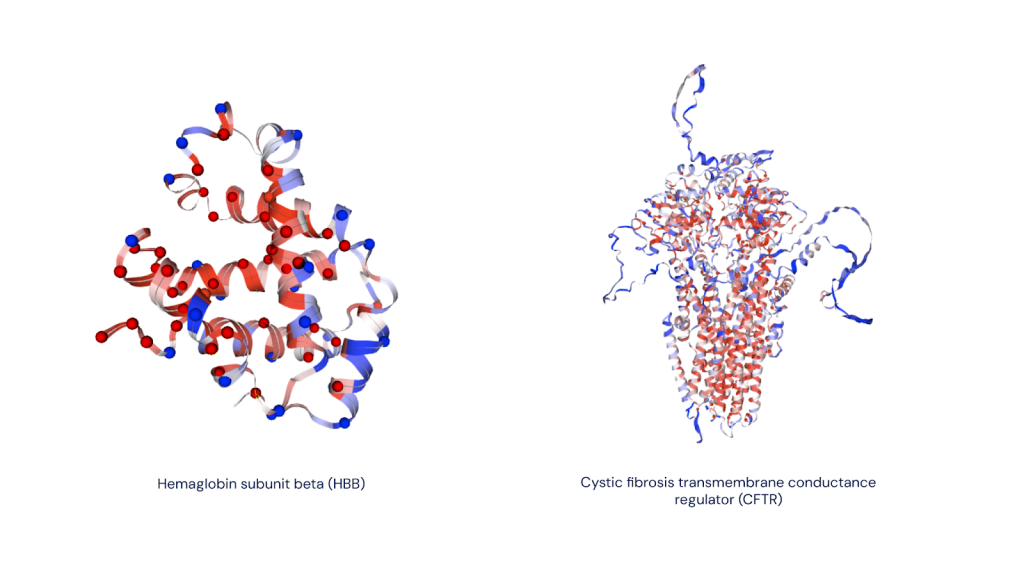
Accélérer la recherche sur les maladies génétiques
Une étape clé dans la traduction de ces recherches consiste à collaborer avec la communauté scientifique. Nous travaillons en partenariat avec Genomics England pour explorer comment ces prédictions pourraient aider à étudier la génétique des maladies rares. Genomics England a croisé les résultats d’AlphaMissense avec des données sur la pathogénicité des variantes précédemment regroupées avec des participants humains. Leur évaluation a confirmé que nos prédictions sont exactes et cohérentes, fournissant ainsi une autre référence concrète pour AlphaMissense.
Bien que nos prédictions ne soient pas conçues pour être utilisées directement en clinique – et doivent être interprétées avec d’autres sources de preuves – ces travaux ont le potentiel d’améliorer le diagnostic de maladies génétiques rares et d’aider à découvrir de nouveaux gènes à l’origine de maladies.
À terme, nous espérons qu’AlphaMissense, associé à d’autres outils, permettra aux chercheurs de mieux comprendre les maladies et de développer de nouveaux traitements salvateurs.

AlphaMissense pathogenicity prediction.
AlphaMissense takes as input a missense variant and predicts its pathogenicity. We fine-tuned AlphaFold on human and primate variant population frequency data and calibrated the confidence on known disease variants. AlphaMissense predicts the probability of a missense variant being pathogenic and classifies it as either likely benign, likely pathogenic, or uncertain. We provide predictions for all possible human missense variants as a resource for the community.
pour télécharger les base dedonnées :
L’article scientifique détaillé :
https://www.science.org/stoken/author-tokens/ST-1429/full#sec-2